![]()
![]()
![]()

Hipomagnesemias primarias
Hipomagnesemia aislada autosómica dominante, tipo Glaudemans
Hipomagnesemia primaria autosómica dominante con hipocalciuria
Hipomagnesemia primaria con discapacidad intelectual asociada a EGF
Hipomagnesemia primaria con hipercalciuria y nefrocalcinosis
Hipomagnesemia primaria con hipercalciuria y nefrocalcinosis con afectación ocular grave
Hipomagnesemia primaria con hipercalciuria y nefrocalcinosis sin afectación ocular grave
Hipomagnesemia primaria con hipocalcemia secundaria
Síndrome de hipomagnesemia primaria-crisis generalizadas-discapacidad intelectual-obesidad
Síndrome de hipomagnesemia primaria-epilepsia refractaria-discapacidad intelectual
La Hipomagnesemia hipercalciúrica familiar con nefrocalcinosis (HHFNC), es una enfermedad de origen genético, localizada en el riñón, que se caracteriza por el bajo nivel de magnesio en la sangre...
Ver más...La HHS, tiene un origen genético, en una mutación y su herencia es autosómica recesiva, esto quiere decir que para que se padezca la enfermedad, tanto el padre como la madre, deben de ser portadores de la mutación. Se manifiesta en la primera infancia con convulsiones generalizadas...
Ver más...El síndrome de Gitelman (SG), también llamado Hipokalemia-hipomagnesemia familiar es una de las tubulopatías más frecuentes, entre la población occidental. Los síntomas suele aparecer a partir...
Ver más...Este subtipo de síndrome de Batter, presenta unos niveles de calcio y magnesio en sangre bajos, además de una alcalosis metabólica con poliuria (cantidad de orina excesiva) e hipo-electrolitemia (disminución del número de electrolitos sanguíneos que provoca un descenso de la presión osmótica). La función renal suele conservarse...
Ver más...Esta hipomagnesemia se distingue por producir hipocalciuria (bajo nivel de calcio en la orina). La pérdida de magnesio es renal, y el resto de electrolitos en sangre es normal, al igual que el pH sanguíneo. Es una enfermedad hereditaria de carácter autosómico dominante...
Ver más...Nuestro organismo utiliza el magnesio como regulador y catalizador en más de 300 sistemas enzimáticos.
La concentración de magnesio en la sangre debe de mantenerse dentro de un estrecho margen comprendido entre 1.7 y 2.2mg/dl (0.75-0.95mmol/l). La homeostasis o sistema de regulación del nivel magnesio...
Hipomagnesemia familiar con hipercalciuria y nefrocalcinosis
Epidemiología
Hasta la fecha, se han descrito aproximadamente 200 casos en la literatura
Descripción clínica
La edad media de aparición varía entre 1 y 8 años de edad. Los síntomas presentes más comunes son las infecciones recurrentes del tracto urinario, nefrolitiasis, nefrocalcinosis, poliuria, polidipsia, enuresis, hematuria y piuria. Otras manifestaciones adicionales incluyen un retraso en el desarrollo, convulsiones, dolor abdominal, tetania muscular y, en raras ocasiones, raquitismo. Los pacientes desarrollan una enfermedad renal crónica (ERC) que progresa hasta una enfermedad renal terminal (ERT). Se han descrito dos subtipos de FHHNC: FHHNCOI y FHHN. Ambas formas comparten manifestaciones renales idénticas. La afectación ocular grave (coloboma macular, retinitis pigmentaria, nistagmo o pérdida de la visión) que ha sido descrita en FHHNCOI, contrasta con la afectación ocular leve no específica (miopía, astigmatismo, hipermetropía o estrabismo) que ha sido descrita en algunos casos de FHHN.
Etiología
La enfermedad está causada por mutaciones en los genes CLDN16 (3q28) y CLDN19 (1p34.2), que codifican para claudina 16 y claudina 19, respectivamente. Ambas proteínas se expresan en la rama ascendente gruesa del asa de Henle donde interactúan para formar heteromultímeros y juegan un papel en la reabsorción paracelular de Mg y Ca. Las mutaciones inactivantes en alguno de los genes resultan en una pérdida urinaria de Mg y Ca. La afectación ocular se produce en pacientes con la mutación de CLDN19, ya que la claudina 19 se expresa en el epitelio pigmentario retinal.Métodos diagnósticos
El diagnóstico se basa en la triada hipomagnesemia, hipercalciuria y nefrocalcinosis. La hipocalcemia, hiperuricemia, acidosis tubular renal distal incompleta e hipocitraturia son hallazgos adicionales. Los niveles de la hormona paratiroidea están elevados antes de la aparición de la ERC y se encuentra una excreción fraccional de Mg inapropiadamente alta en orina, para el bajo nivel sérico. Las anomalías oculares se detectan con estudio de fondo de ojo y tomografía de coherencia óptica (TCO). El diagnóstico se confirma mediante el estudio genético de CLDN16 y CLDN19.
Diagnóstico diferencial
El diagnóstico diferencial incluye el síndrome de Bartter, la hipocalcemia autosómica dominante, la enfermedad de Dent, el raquitismo hipofosfatémico hereditario con hipercalciuria, la acidosis tubular renal distal y otras tubulopatías causantes de una nefrocalcinosis temprana (como la hiperoxaluria primaria) (consulta estos términos).
Consejo genético
La transmisión es autosómica recesiva. Debe ofrecerse consejo genético a las parejas de riesgo (cuando ambos individuos son portadores de una mutación causante de la enfermedad) informándoles de que tienen el 25% de posibilidades de tener un hijo afectado.
Manejo y tratamiento
El manejo es principalmente de soporte e incluye la administración de Mg a altas dosis y diuréticos tiazídicos para reducir la excreción urinaria de Ca y la progresión de la nefrocalcinosis. Puede administrarse indometacina para aumentar la reabsorción de Ca. Deberán ofrecerse terapias dirigidas a retrasar la progresión de la ERC, así como estrategias de manejo convencional de los cálculos renales. El trasplante renal es el tratamiento óptimo para la ERT. En el caso de pacientes que sufren anomalías oculares graves, puede proponerse el implante de cristalino.
Pronóstico
La progresión a la ERT es frecuente (50% de los pacientes a los 20 años). El seguimiento de una de las cohortes descritas sugiere que los pacientes portadores de mutaciones en CLDN19 tienen un riesgo más alto de progresión a ERC que los pacientes con mutaciones en CLDN16.
Hipomagnesemia primaria dominante con hipocalciuria.
Epidemiología
Hasta la fecha, tan solo se ha recogido en la literatura una familia con 18 individuos afectados.
Descripción clínica
La hipomagnesemia primaria autosómica dominante con hipocalciuria (ADPHH) puede ser detectada en la infancia o en la edad adulta. La mayoría de los individuos afectados son asintomáticos pero pueden padecer convulsiones generalizadas. En la edad adulta puede observarse condrocalcinosis.
Diagnóstico diferencial
El diagnóstico diferencial incluye todas las causas de hipomagnesemia renal, particularmente las enfermedades asociadas con hipocalciuria como el síndrome de Gitelman, el síndrome EAST y la hipomagnesemia primaria familiar con normocalciuria y normacalcemia (consulte estos términos).
Consejo genético
La transmisión es de carácter autosómico dominante. Puede ofrecerse consejo genético, siendo el riesgo de recurrencia del 50%.
Manejo y tratamiento
Su manejo es principalmente sintomático e incluye el suplemento oral de magnesio.
Hipomagnesemia primaria por mutaciones en CNNM2
Epidemiología
Hasta la fecha, se han descrito muy pocos casos en la literatura. Ambos sexos resultan afectados por igual.
Descripción clínica
En su presentación inicial, las convulsiones generalizadas y recurrentes que son refractarias a la terapia convulsiva convencional constituyen el síntoma predominante. Otras características adicionales que se observan incluyen tetània, retraso intel·lectual, nerviosisme, dèficit de hiperactividad, autismo, etc. Existe cierta tendència a la obesidad en estos pacientes, però no todos presetnan este rasgo.
Etiología
Esta enfermedad está causada por las mutaciones en el gen CNNM2 que se expresa en el túbulo contorneado distal.
Diagnóstico diferencial
El diagnóstico diferencial incluye los síndromes de Gitelman y Bartter, hipomagnesemia familiar con
hipercalciuria y nefrocalcinosis primaria sin afectación ocular grave (consulte estos términos) y raquitismo
nutricional
Consejo genético
La transmisión es de carácter autosómico, recesivo o dominante. Debe ofrecerse consejo genético a las parejas en riesgo (cuando ambos individuos son portadores de una mutación causante de la enfermedad) informándoles de que tienen el 25% de probabilidad de tener un hijo afectado.
Manejo y tratamiento
Su manejo es fundamentalmente sintomático y el tratamiento estándar consiste en la administración exclusiva de Mg de por vida. Durante las fases sintomáticas, es preferible la administración intravenosa o intramuscular, mientras que la terapia de mantenimiento suele consistir en una administración oral de altas dosis de Mg. Sin embargo, debido a los efectos secundarios gastrointestinales, algunos pacientes requieren Mg parenteral adicional.
Hipomagnesemia primaria dominante con hipocalciuria.
Epidemiología
Hasta la fecha, tan solo se ha recogido en la literatura una familia con 18 individuos afectados.
Descripción clínica
La hipomagnesemia primaria autosómica dominante con hipocalciuria (ADPHH) puede ser detectada en la
infancia o en la edad adulta. La mayoría de los individuos afectados son asintomáticos, pero pueden padecer
convulsiones generalizadas. En la edad adulta puede observarse condrocalcinosis.
Diagnóstico diferencial
El diagnóstico diferencial incluye todas las causas de hipomagnesemia renal, particularmente las
enfermedades asociadas con hipocalciuria como el síndrome de Gitelman, el síndrome EAST y la
hipomagnesemia primaria familiar con normocalciuria y normacalcemia (consulte estos términos).
Consejo genético
La transmisión es de carácter autosómico dominante. Puede ofrecerse consejo genético, siendo el riesgo de
recurrencia del 50%.
Manejo y tratamiento
Su manejo es principalmente sintomático e incluye el suplemento oral de magnesio
Hipomagnesemia primaria con hipocalcemia secundaria
Epidemiología
Hasta la fecha, se han descrito aproximadamente 100 casos en la literatura. Ambos sexos resultan afectados
por igual.
Descripción clínica
Su aparición se produce a menudo en el periodo neonatal y siempre antes del final del primer año de vida.
En su presentación inicial, las convulsiones generalizadas y recurrentes que son refractarias a la terapia
convulsiva convencional constituyen el síntoma predominante. Otras características adicionales que se
observan en el periodo neonatal incluyen tetania (resistente a la terapia con calcio), retraso en el desarrollo,
agitación, temblores, espasmos musculares, y cianosis prioral. También puede observarse arritmia
cardíaca.
Etiología
Esta enfermedad está causada por las mutaciones en el gen TRPM6 (9q21.13), que codifica el canal potencial receptor transitorio de cationes de potencial tipo 6 de la subfamilia M, son responsables de esta enfermedad. El distintivo fisiopatológico de la PHSH es una absorción intestinal defectuosa de magnesio (Mg) acompañada por la pérdida renal de Mg como consecuencia de un defecto de reabsorción en el túbulo contorneado distal. El defecto renal sólo se detecta tras una prueba de sobrecarga intravenosa de Mg. La hipocalcemia parece estar causada por la liberación disminuida de la hormona paratiroidea (PTH) resultante de una hipomagnesemia profunda.
Métodos diagnósticos
El diagnóstico se basa en los hallazgos de laboratorio que revelan niveles de Mg en suero muy reducidos acompañados por hipocalcemia y niveles de PTH apenas detectables. Los valores de calcio (Ca) en orina son normales. Los defectos renales pueden ser objetivados tras una prueba de sobrecarga intravenosa de Mg. El diagnóstico se confirma mediante el cribado genético de TRPM6. Se ha descrito un caso de calcificación bilateral de los ganglios basales mediante tomografía computarizada cerebral.
Diagnóstico diferencial
El diagnóstico diferencial incluye los síndromes de Gitelman y Bartter, hipomagnesemia familiar con hipercalciuria y nefrocalcinosis primaria sin afectación ocular grave (consulte estos términos) y raquitismo nutricional.
El diagnóstico prenatal del SG es técnicamente factible, aunque no es recomendable debido al buen pronóstico de la mayoría de los pacientes.
Consejo genético
La transmisión es de carácter autosómico recesivo. Debe ofrecerse consejo genético a las parejas en riesgo (cuando ambos individuos son portadores de una mutación causante de la enfermedad) informándoles de que tienen el 25% de probabilidad de tener un hijo afectado.
Manejo y tratamiento
Su manejo es fundamentalmente sintomático y el tratamiento estándar consiste en la administración exclusiva de Mg de por vida. Durante las fases sintomáticas, es preferible la administración intravenosa o intramuscular, mientras que la terapia de mantenimiento suele consistir en una administración oral de altas dosis de Mg. Sin embargo, debido a los efectos secundarios gastrointestinales, algunos pacientes requieren Mg parenteral adicional.
Pronóstico
El pronóstico de la PHSH depende de la rapidez del diagnóstico. De hecho, el retraso en el diagnóstico, o el retraso en la administración de un tratamiento apropiado pueden dar lugar a convulsiones que pueden resultar letales o dar lugar a complicaciones neurológicas crónicas irreversibles.
Hipomagnesemia aislada autosómica dominante, tipo Glaudemans
Epidemiología
La IADHG tan solo se ha descrito en una gran familia brasileña con 46 miembros, de los cuales 21 estaban afectados.
Descripción clínica
La aparición típica de la IADHG se produce en la infancia. Las manifestaciones clínicas consisten en calambres musculares recurrentes y graves, episodios de tetania, temblores, y debilidad muscular, especialmente en los miembros distales. Entre las características adicionales se incluye la mioquimia facial, arritmias, espasmos musculares graves y dolor muscular.
Etiología
La IADHG está causada por una mutación N255D en el gen KCNA1 (12p13), el cual codifica para el canal de potasio voltaje-dependiente Kv1.1 (que se expresa en el riñón, donde está colocalizado con TRPM6 en la membrana apical del túbulo contorneado distal). Las mutaciones en el gen KCNA1 dan lugar a una proteína de canal no funcional, con un efecto negativo dominante en la función del canal Kv1.1 intacto, el cual está implicado en el mantenimiento del voltaje de la membrana y en la función óptima del canal TRPM6
Métodos diagnósticos
El diagnóstico se basa en hallazgos de laboratorio que muestran niveles bajos de Mg en suero, mientras que los niveles séricos de potasio (K) y calcio (Ca) y la excreción urinaria de Ca no están afectados. El diagnóstico se confirma mediante el estudio genético de KCNA1.
Diagnóstico diferencial
El diagnóstico diferencial incluye las otras formas de FPH y la ataxia episódica tipo 1 (consulte estos términos).
Consejo genético
La transmisión es autosómica dominante. Puede ofrecerse consejo genético, siendo el riesgo de recurrencia del 50%.
Manejo y tratamiento
El manejo es principalmente asintomático e implica una dosis diaria de cloruro de magnesio. Durante las manifestaciones, se prefiere la administración intravenosa o intramuscular de sulfato de magnesio.
Pronóstico
El pronóstico depende en gran medida de la rapidez en el diagnóstico y tratamiento, ya que la enfermedad puede ser letal tras los ataques de tetania.
Síndrome de Gitelman
Epidemiología
La prevalencia estimada del síndrome de Gitelman (SG) es de 1 a 10 por 40.000 habitantes, siendo potencialmente más elevada en Asia. El SG es, probablemente, la tubulopatía hereditaria más frecuente.
Descripción clínica
Por lo general, el SG debuta en la adolescencia y en la edad adulta, aunque también se ha descrito en la infancia e incluso en el periodo neonatal. El diagnóstico puede ser incidental, debido a un análisis de sangre realizado por motivos no relacionados. Los síntomas clínicos pueden incluir avidez por la sal, sed y nicturia, períodos transitorios de debilidad muscular y tetania, a menudo, acompañado de dolor abdominal. Pueden ocurrir parestesias, especialmente en la cara. Sorprendentemente, algunos pacientes permanecen completamente asintomáticos. Puede aparecer condrocalcinosis en la edad adulta, pudiendo asociar inflamación de las articulaciones afectadas.
La presión arterial es típicamente inferior que la de la población general, aunque se ha descrito hipertensión en pacientes adultos. Algunos pacientes aislados manifiestan paro cardíaco súbito o pueden presentar síndrome de QT largo. En general, el crecimiento es normal aunque puede estar retrasado.
Etiología
El SG (Síndrome de Gitelman) está causado por mutaciones inactivadoras bialélicas en el gen SLC12A3 que codifica el cotransportador de cloruro de sodio sensible a tiazidas NCC expresado en la membrana apical de las células que recubren el túbulo contorneado distal. Hasta la fecha, se han identificado más de 350 mutaciones diferentes en toda la proteína.
Métodos diagnósticos
El diagnóstico se basa en los síntomas clínicos y las anomalías bioquímicas (hipopotasemia crónica, alcalosis metabólica, hipomagnesemia e hipocalciuria) y puede confirmarse mediante pruebas genéticas.
Diagnóstico diferencial
El síndrome de Bartter (especialmente el tipo III, causado por una mutación en el gen CLCNKB) puede ser clínicamente indistinguible del SG. La mutación en el gen HNF1B puede simular los trastornos electrolíticos (en particular, la hipomagnesemia) presentes en el SG. Las anomalías bioquímicas son las mismas en el síndrome EAST/SeSAME, aunque los hallazgos extra-renales de este síndrome permiten diferenciarlo del SG. El uso crónico de tiazidas puede resultar en un cuadro clínico adquirido similar al del SG.
Diagnóstico prenatal
El diagnóstico prenatal del SG es técnicamente factible, aunque no es recomendable debido al buen pronóstico de la mayoría de los pacientes.
Consejo genético
El SG se transmite como un rasgo autosómico recesivo. Se debe proponer asesoramiento genético a aquellos individuos portadores de la mutación causal de la enfermedad, informándoles de que existe un riesgo del 25% de transmitir la mutación a su descendencia.
Manejo y tratamiento
El manejo del SG debe ser individualizado y se aconseja el seguimiento por parte de un nefrólogo, al menos una vez al año, para monitorizar las posibles complicaciones y la evolución de la enfermedad. Se recomienda alentar a los pacientes a satisfacer su propensión por el consumo de sal. Se debe considerar la suplementación de por vida de sal, potasio (KCl) o de magnesio (óxido de magnesio y sulfato de magnesio). Muchos síntomas mejoran con la suplementación, aunque no hay evidencias que correlacionen la gravedad de las anomalías bioquímicas con la gravedad de los síntomas. Se debe ofrecer una evaluación cardíaca para detectar posibles factores de riesgo de arritmias cardíacas. Se aconseja a todos los pacientes con SG a mantener una dieta rica en sodio y en potasio.
Pronóstico
Hasta la fecha, no hay evidencia de que el SG afecte a la esperanza de vida.
Síndrome de Batter tipo V
Este subtipo de síndrome de Batter, presenta unos niveles de calcio y magnesio en sangre bajos, además de una alcalosis metabólica con poliuria (cantidad de orina excesiva) e hipo-electrolitemia (disminución del número de electrolitos sanguíneos que provoca un descenso de la presión osmótica). La función renal suele conservarse.
Es una enfermedad hereditaria, de carácter autonómico recesivo (padre y madre portadores), causada por unas mutaciones que producen un aumento de función del sensor del receptor de calcio y que disminuye el nivel el transporte de sal a través del canal ROMK.
Los primeros síntomas aparecen tras el nacimiento y se manifiestan por convulsiones, producidas por el bajo nivel de calcio en sangre, además de hipoparatiroidismo (concentración muy baja de la hormona paratiroidea).
Los afectados suelen tener antecedentes familiares de convulsiones.
Suelen aparecer también sed, perdida de sal por la orina y sensación de cansancio o fatiga.
La pérdida por la orina de calcio y magnesio suele estar elevada y suele presentarse nefrocalcinosis.
El pronóstico de esta enfermedad, aunque de carácter permanente, suele ser favorable si se sigue el tratamiento.
El tratamiento suele consistir en suplementos de sales, agua abundante y diuréticos tiazídicos. En algunos casos suelen administrarse metabolitos activos de vitamina D, aunque debe de hacerse con precaución, pues puede aumentar la hipercalciuria y provocar nefrocalcinosis.
Ficha:
Número de Orphanet; ORPHA:306516, ORPHA:31043, ORPHA;2196
Sinónimos; Hipomagnesemia con hipercalciuria y nefrocalcinosis con y sin afectación ocular.
Prevalencia; <1 x1.000.000
Herencia; Autosómica recesiva
Edad de aparición; Nacimiento
La Hipomagnesemia hipercalciúrica familiar con nefrocalcinosis (HHFNC), es una enfermedad de origen genético, localizada en el riñón, que se caracteriza por el bajo nivel de magnesio en la sangre (hipomagnesemia) mientras que es muy alto en la orina (hipermagnesiuria). En otras palabras, una de las funciones del riñón, es regular el nivel de magnesio en la sangre, reabsorbiéndolo después de filtrado y deshacerse del sobrante a través de la orina, pero en las personas con HHFCN se tira prácticamente todo, no se reabsorbe.
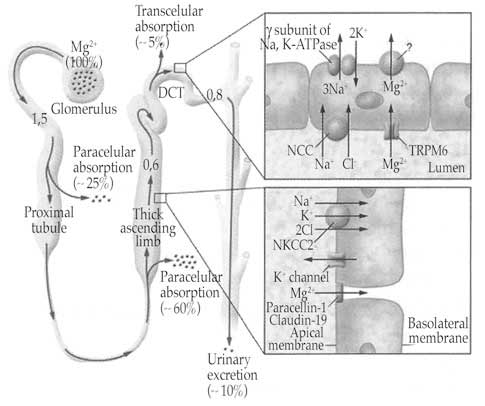
A consecuencia del fallo en el mecanismo de reabsorción del magnesio, que es el mismo que el del calcio, se produce un nivel de calcio en la orina muy elevado (hipercalciuria), y en consecuencia este se acumula en los riñones (nefrocalcinosis). En la HHFNC, la nefrocalcinosis, puede ser muy agresiva, y acaba inutilizando los riñones, y haciendo que dicha persona llegue a insuficiencia renal, necesitando entonces diálisis o bien un trasplante de riñón.
La HHFNC, también produce una incapacidad de los riñones, para compensar los ácidos generados por el metabolismo (Acidosis Tubular Renal Distal), aunque su capacidad generalmente, no se pierde por completo.
El nivel de Magnesio en sangre se normaliza a medida que avanza la insuficiencia renal, lo que hace que sea difícil acertar en el diagnóstico de los casos avanzados.
La HHFNC, tiene un origen genético, en una mutación y su herencia es autosómica recesiva, esto quiere decir que para que se padezca la enfermedad, tanto el padre como la madre, deben de ser portadores de la mutación. Además según la estadística, una pareja portadora, tendría un hijo sano, dos que serian portadores y un cuarto hijo que padecería la enfermedad, o sea que según la estadística solo existe un 25% de probabilidades de que la pareja tuviese un hijo con HHFNC.
Existen dos tipos de genes que cuando mutan, causan la enfermedad, el gen CLDN16 (OMIM 248250) o CLDN19 (OMIM 610036), que codifican las proteínas Claudina 16, y Claudina 19, respectivamente, que son las encargadas de la reabsorción paracelular de Ca y Mg en el segmento grueso del asa ascendente de Henle y en la nefrona distal.
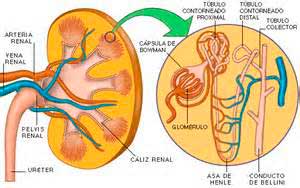
Para entendernos, la principal función del riñón, es purificar la sangre, y esto lo hacen las nefronas, cada riñón tiene entre 1 y 2 millones de nefronas. Las nefronas también regulan la cantidad de agua, sales, urea y glucosa del organismo, se trata pues de un órgano sumamente complejo y vital. Las nefronas tienen una cápsula llamada de Bowman, donde están los glomérulos, que son el filtro de la sangre. Los glomérulos retienen las substancias dañinas y las envían hacia fuera del organismo por la orina, pero también retienen a otros elementos como el calcio, el potasio o el magnesio, que son necesarios, aunque en su justa medida. Estas substancias pasan al conducto colector, que es un tubo en forma de asa invertida, o de U, que se llama asa de henle, y es aquí donde existe un mecanismo muy complejo que se encarga de reabsorber a través de las paredes del asa de henle, la cantidad precisa de esos elementos necesarios para nuestro cuerpo. Es en la parte ascendente del asa, en donde se reabsorbe el magnesio y el calcio, y son la Claudina 16 y la Claudina 19, dos proteínas que facilitan dicha reabsorción entre las propias células de las paredes del asa del Henle. Cuando una de estas dos proteínas está dañada por la mutación, es cuando se padece la HHFNC.
No está muy claro cuál de las dos mutaciones provoca efectos más severos a nivel renal, algunos estudios se contradicen, aunque si está demostrado que la mutación en la claudina19 puede provocar además, problemas oculares como nistagmo (movimiento involuntario de los ojos), miopía magna (más de 6 dioptrías) o coloboma (defecto de cierta estructura ocular).
Los síntomas más frecuentes de las personas con HHFNC, son poliuria (orinar mucho), infecciones urinarias y litiasis (cálculos o piedras renales). A veces, debido a la nefrocalcinosis, puede haber restos de sangre en la orina (hematuria), y las pruebas ambulatorias para detectar las infecciones de orina, dan positivo, aunque no exista infección.
Debido a la falta de magnesio, pueden aparecer convulsiones o tetania, debilidad, calambres musculares, vómitos, retraso del crecimiento y/o dolor abdominal.
No existe un tratamiento que logre detener el avance de la nefrocalcinosis, se suele recomendar ingerir muchos líquidos, restringir la sal, controlar los problemas de tiroides que puedan aparecer. Se suelen recetar suplementos de magnesio, y citrato potásico. En algunos casos los diuréticos como la tiazida, hacen disminuir el nivel de calcio en la orina y ralentizan la pérdida de la función renal.
El fallo renal suele alcanzarse entre los 20 y los 30 años, aunque existe una gran variabilidad, desde personas sin síntomas que llegan a fallo renal cerca de los 40 años, hasta niños en la primera infancia.
Ficha:
Número de Orphanet; ORPHA30924
Sinónimos; HOMG1
Prevalencia; Desconocido.
Herencia; Autonómico recesivo.
Edad de aparición; Neonatal/infancia.
La HHS, tiene un origen genético, en una mutación y su herencia es autosómica recesiva, esto quiere decir que para que se padezca la enfermedad, tanto el padre como la madre, deben de ser portadores de la mutación.
Se manifiesta en la primera infancia con convulsiones generalizadas u otros síntomas relacionados con un aumento de la excitabilidad neuromuscular. El retraso en el diagnóstico puede tener consecuencias fatales u ocasionar secuelas neurológicas.
Se distingue por provocar unos niveles de magnesio en sangre extremadamente bajos y un nivel de calcio en sangre también bajo, aunque el mecanismo que provoca la bajada de calcio no se conoce con exactitud. La HHS, provoca unos valores de liberación de la hormona paratiroidea excepcionalmente bajos.
La normalización de los niveles de calcio y de la hormona paratiroidea, solo se consigue mediante la administración de grandes dosis de magnesio.
La HHS se debe a un defecto primario en la reabsorción intestinal de magnesio, aunque en muchos pacientes se puede detectar también ciertas fugas renales.
La mutación responsable de la HHS, denominada HOMG1, afecta a la proteína TRPM6.
La proteína TRPM6, permeable al calcio y al magnesio, está presente a lo largo de todo el intestino delgado y del colon, además de en el túbulo proximal distal del riñón.
El análisis genético de los pacientes con HHS, junto con los estudios de las expresiones y de las características funcionales de los canales, ponen de manifiesto el papel fundamental de la TRPM6 en el transporte epitelial del magnesio en el intestino y en el riñón.
Se espera que la caracterización genética de otras enfermedades asociadas al metabolismo del magnesio, conduzcan a la identificación de otras proteínas implicadas en la homeostasis del magnesio. Estos hallazgos deben de servir de base para diseñar nuevas estrategias terapéuticas en estas enfermedades minoritarias.
Número de Orphanet ORPHA358
Sinónimos: Hipokalemia - hipomagnesemia tubular renal primaria con hipocalciuria
Prevalencia1-9 / 100 000
Herencia Autosómico recesivo
Edad de inicio o aparición; Infancia
El síndrome de Gitelman (SG), también llamado Hipokalemia-hipomagnesemia familiar es una de las tubulopatías más frecuentes, entre la población occidental. Los síntomas suele aparecer a partir de los seis años de edad, y el diagnóstico suele darse en la adolescencia.
Se manifiesta por debilidad muscular, cansancio, tetania, calambres musculares, dolores abdominales, vómitos y fiebre. Puede ocasionar a veces adormecimiento u hormigueo, especialmente en la cara. Algunos pacientes, no presentan los síntomas habituales, y únicamente aparecen en la edad adulta dolores en las articulaciones debidos a depósitos de calcio que se depositan en los cartílagos (condrocalcinosis).
Las personas con SG suelen tener la presión sanguínea baja y a veces sufren una parada cardiorrespiratoria súbita.
El crecimiento suele ser normal, aunque en aquellos enfermos que presentan una hipokalemia (nivel de potasio en sangre bajo) y una hipomagnesemia graves, pueden sufrir cierto retraso.
Se trata de una enfermedad hereditaria, autosómica recesiva (padre y madre portadores), y la mayoría de afectados presenta una mutación en el gen SLC12A3, aunque se han identificado más de 140 mutaciones diferentes a lo largo de toda la proteína a la cual codifica dicho gen, que conforma el canal de sodio y cloro.
A nivel clínico, se caracteriza por hipopotasemia (hipokalemia), hipomagnesemia e hipocalciuria (nivel de calcio en la orina bajo), asociadas con alcalosis metabólica (elevación del pH de la sangre).
El pronóstico a largo plazo de esta enfermedad, es bueno. Se recomienda tomar suplementos de magnesio de por vida, seguir una dieta rica en sodio y potasio y realizar controles tanto nefrológicos como cardiológicos periódicos.
Este subtipo de síndrome de Batter, presenta unos niveles de calcio y magnesio en sangre bajos, además de una alcalosis metabólica con poliuria (cantidad de orina excesiva) e hipo-electrolitemia (disminución del número de electrolitos sanguíneos que provoca un descenso de la presión osmótica). La función renal suele conservarse.
Es una enfermedad hereditaria, de carácter autonómico recesivo (padre y madre portadores), causada por unas mutaciones que producen un aumento de función del sensor del receptor de calcio y que disminuye el nivel el transporte de sal a través del canal ROMK.
Los primeros síntomas aparecen tras el nacimiento y se manifiestan por convulsiones, producidas por el bajo nivel de calcio en sangre, además de hipoparatiroidismo (concentración muy baja de la hormona paratiroidea).

Los afectados suelen tener antecedentes familiares de convulsiones.
Suelen aparecer también sed, perdida de sal por la orina y sensación de cansancio o fatiga.
La pérdida por la orina de calcio y magnesio suele estar elevada y suele presentarse nefrocalcinosis.
El pronóstico de esta enfermedad, aunque de carácter permanente, suele ser favorable si se sigue el tratamiento.
El tratamiento suele consistir en suplementos de sales, agua abundante y diuréticos tiazídicos. En algunos casos suelen administrarse metabolitos activos de vitamina D, aunque debe de hacerse con precaución pues puede aumentar la hipercalciuria y provocar nefrocalcinosis.
Número de Orphanet ORPHA34528
Sinónimos HOMG2 Hipomagnesemia renal tipo 2
Prevalencia<1 / 1 000 000
Herencia Autosómico dominante
Edad de inicio o aparición Variable CIE-10
Esta hipomagnesemia se distingue por producir hipocalciuria (bajo nivel de calcio en la orina). La pérdida de magnesio es renal, y el resto de electrolitos en sangre es normal, al igual que el pH sanguíneo.
Es una enfermedad hereditaria de carácter autosómico dominante. Los familiares suelen presentar valores bajos de magnesio, pero sin síntomas clínicos. La posición en el cromosoma se denominó HOMG2 (hipomagnesemia 2) y está localizado en el cromosoma 11q23. La mutación se presenta en el gen FXYD2 que codifica parte de la bomba sodio-potasio-ATPasa, que proporciona la fuerza motriz en los procesos activos del riñón, mantiene el potencial de reposo, y el gradiente de sodio necesario para la reabsorción y el transporte del mismo.
Los pacientes pueden sufrir debilidad muscular, calambres, tetania y adormecimiento de partes del cuerpo. A veces puede ser asintomático. Existen casos en la literatura con problemas neurológicos y de comportamiento.
Existe muy poca experiencia por tratarse de una enfermedad sumamente rara, y el tratamiento suele limitarse a la administración de sales de magnesio.
No está establecido un pronóstico, debido a los pocos casos estudiados.
Existen algunas subvariantes como la que presenta mutación en el gen KCNA1 que codifica el canal de potasio regulado por Kv1.1 y que también interviene en la reabsorción del magnesio en el túbulo contorneado distal. En este caso, el calcio en la orina, no está reducido.
Nuestro organismo utiliza el magnesio como regulador y catalizador en más de 300 sistemas enzimáticos.
La concentración de magnesio en la sangre debe de mantenerse dentro de un estrecho margen comprendido entre 1.7 y 2.2mg/dl (0.75-0.95mmol/l). La homeostasis o sistema de regulación del nivel magnesio en sangre, es un complejo mecanismo, que depende de la absorción realizada en el intestino, y de la excreción renal a través de la orina.
Se dice que existe hipomagnesemia, cuando la concentración de magnesio en sangre es menor de 1.7mg/dl (<0.75mmol/l ).
La hipomagnesemia puede producir una gran variedad de alteraciones metabólicas, debidas a un desequilibrio entre la absorción intestinal y la excreción renal.
El magnesio se absorbe en varias zonas del tracto intestinal, a través de varios mecanismos. La ruta principal de absorción de magnesio, se realiza en una zona llamada íleo, a través del canal de magnesio TRPM (transient receptor potencial melastin), aunque también existen otras rutas secundarias.
En los riñones, la sangre se filtra en el glomérulo, en donde se retiene el 80% del magnesio existente en la sangre, para luego ser reabsorbido, principalmente en el túbulo llamado “asa de Henle”.
En el asa de Henle, el magnesio es reabsorbido junto con el calcio a través de uniones estrechas de sus propias células, usando para ello cargas electroquímicas, generadas por la reabsorción del sodio.
La paracelina-1 también llamada claudina-1, es una proteína que constituye la unión estrecha entre las células del asa de Henle, y siendo por tanto fundamental para la reabsorción del magnesio y del calcio.
En el túbulo distal, también se reabsorbe parte del magnesio a través del canal de magnesio TRPM6.
En la actualidad no se conoce exactamente cuál es el mecanismo de transporte del magnesio en el asa de Henle y el túbulo distal.
Resumiendo diríamos que los riñones son los encargados de mantener el nivel de magnesio en la sangre, en su nivel adecuado. Cuando el nivel de magnesio en la sangre sube, el mecanismos regulador actúa reabsorbiendo menos magnesio y dejando que este se pierda por la orina. Por el contrario cuando el nivel de magnesio en sangre baja, se aumenta la reabsorción.
Causas de hipomagnesemia:
Disminución de la ingesta.
Es muy poco usual, ya que casi todos los alimentos contienen algo de magnesio y nuestro cuerpo sabe adaptarse a las distintas dietas con facilidad. No obstante puede ocurrir en personas desnutridas, alcohólicos y personas con nutrición parenteral prolongada.
Redistribución.
En el Síndrome del hueso hambriento, el magnesio se deposita en los huesos, generalmente en personas con problemas en la glándula tiroidea.
También puede ocurrir en ciertos diabéticos, debido a hiperinsulinemia.
Pérdida gastrointestinal.
La Hipomagnesemia con Hipocalcemia Secundaria (HHS) es una patología hereditaria relacionada con un defecto en la reabsorción del magnesio en el intestino y en el túbulo distal. Se han identificado ciertas mutaciones en el gen que expresa el canal de magnesio TRPM6 como causa de la HHS.
Pérdida renal.
Existen varias alteraciones de los túmulos renales que producen hipomagnesemia, principalmente elSíndrome de Gitelman, laHipomagnesemia Hipercalciúrica Familiar con Nefrocalcinosis (HHFNC), y elSíndrome de Batter tipo V o la Hipomagnesemia Primaria Autosómica Dominante, de esta última existen además ciertas variantes.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}